代谢重编程是癌症的重要标志,它为癌症患者创造了新的治疗机会并推动越来越多的抗癌药物发现。最近,人们越来越认识到生化通路改变通常会诱发癌细胞代谢的弱点。代谢重编程被认为是由代谢酶的遗传改变或致癌基因激活引起的。目前,只鉴定到少数代谢酶基因的异常。在代谢变化中,致癌因子活化的癌细胞是否会表现出代谢偏向性尚不清楚。酪氨酸激酶受体(RTK),如表皮生长因子受体(EGFR)和成纤维细胞生长因子受体(FGFR),是公认的各种癌症恶性生长的致癌因子。迄今为止,RTK基因改变代表了人类癌症中最明确的遗传亚型,特别是在非小细胞肺癌(NSCLC)中。虽然越来越多的证据证明了RTK对代谢网络重编程的影响,但RTK驱动的代谢编程是否导致代谢弱点而具有治疗潜力仍未可知。中科院上海药物所黄敏研究团队和厦门大学生科院林树海教授通过非靶向代谢组学、多种稳定同位素标记的代谢流分析及转录组学等技术阐述了癌症基因型和代谢依赖之间的分子联系,为靶向代谢治疗中的患者分层提供了依据,相关成果发表于《Nature Communications》。(文末有下载原文方式)
RTK的代谢表型重编程
采用药理学抑制剂筛查验证RTK改变导致的癌症代谢弱点。选用癌基因表达异常的15株NSCLC细胞株,包括EGFR突变(L858R,外显子19缺失或外显子21缺失),FGFR1扩增,KRAS突变等,暴露于靶向葡萄糖和谷氨酰胺酶或脂肪酸氧化的代谢抑制剂中。生长抑制率的聚类结果表明,相同基因型的癌细胞具有相似的代谢弱点,特别是对于表现出聚集趋势的FGFR和EGFR异常细胞。为了证实这一发现的临床相关性,作者从TCGA数据库中提取了740例肺腺癌,其中54例患者分别接受了EGFR激活突变(n = 25),FGFR1 /2扩增(n = 15),MET扩增(n = 12),RET融合(n = 2)。在这些样本中,基于KEGG数据库中注释的1498个代谢基因的层次聚类结果表明,EGFR-,FGFR-和RET-激活的肿瘤存在不同的表达模式,提示在RTK驱动的癌症中致癌基因存在显著的代谢表型差异。
为了探究RTK进行代谢网络重编程的偏向性,将致癌基因EGFR(EGFR-L858R-T790M),FGFR1(TEL-FGFR1融合体),MET(TPR-MET融合体)或RET(CCDC6-RET融合体)引入BAF3细胞导致激活RTK信号传导(Fig. 1a),IL3非依赖性细胞生长(Fig. 1b),及对特异性RTK抑制剂的精确敏感性(Fig. 1c),并对这些细胞系的代谢谱进行表征。通过细胞外酸化率(ECAR)和氧消耗率(OCR)可知,RTK活化可以促进糖酵解和氧化磷酸化,但RTK基因型之间存在显著差异(Fig. 1d)。鉴于FGFR基因具有四种亚型,作者还将TEL-FGFR3融合到BAF3细胞中,其导致IL3非依赖性细胞生长和对AZD4547的敏感性。比较分别由FGFR1和FGFR3驱动的BAF3细胞表明,发现两者ECAR和OCR均增强。因为IL3对BAF3细胞模型非常重要,作者测试IL3对细胞代谢表型的影响。正如预期,IL3的缺失导致BAF3细胞中OCR的显著变化,因为这些细胞的存活率依赖于IL3。BAF3-RTK细胞受影响较小,这与IL3对细胞生长的影响相关(Fig. 1b)。
对这些细胞系进行非靶向代谢组学分析,鉴定了124个代谢物,单个代谢物的热图(Fig. 1e)、主成分分析(PCA)(Fig. 1f)、通路富集分析(倍数大于1.5倍; p <0.01)突出了恶性细胞生长所需要的几种代谢途径,尤其是柠檬酸循环(TCA循环),核苷酸生物合成和氨基酸代谢,在FGFR突变细胞中优先激活TCA循环,在BAF3-RET细胞中谷氨酰胺/谷氨酸代谢通路显著增强。
代谢异质性提示增殖细胞营养获取能力的差异。采用同位素光谱分析(ISA)检测13C标记的葡萄糖、谷氨酰胺或棕榈酸在中间代谢物中的掺入,以追踪三种主要的营养源---葡萄糖,谷氨酰胺和脂肪酸的代谢(Fig. 1g)。每个细胞系中13C标记代谢物的热图显示,BAF3-EGFR和BAF3-FGFR1细胞中葡萄糖代谢增强,BAF3-RET细胞中谷氨酰胺分解显著增强,MET扩增细胞没有表现出显著的代谢特征(Fig. 1h)。
那么RTK驱动细胞的代谢变化是否具有代谢依赖性?结果显示,BAF3-EGFR和BAF3-FGFR1细胞的增殖依赖于葡萄糖供应,而BAF3-RET细胞的生长似乎依赖于谷氨酰胺供应(Fig. 1i)。检测9种EGFR突变细胞,4种FGFR1/3扩增细胞和2种RET融合/突变细胞对葡萄糖或谷氨酰胺的生长依赖性,并使用癌症细胞系百科全书(CCLE)数据库注释的无驱动基因改变的7株野生型肺癌细胞(A431除外)作为对照,结果表明,与BAF3-RTK细胞中的观察一致,EGFR和FGFR突变细胞的生长依赖于葡萄糖而不是谷氨酰胺。相反,对照野生型细胞对葡萄糖或谷氨酰胺的依赖程度不同。RET融合/突变细胞依赖谷氨酰胺进行增殖。与该结果一致,使用UK5099(线粒体-丙酮酸盐载体抑制剂)选择性抑制葡萄糖代谢,优先抑制EGFR和FGFR依赖性癌细胞的生长。谷氨酰胺酶抑制剂CB839对RET异常癌细胞的生长表现出更显著的影响。此外,与脂肪酸β-氧化示踪一致(Fig. 1h),使用埃托霉素(ETO)(一种肉毒碱棕榈酰转移酶1A(CPT1A)的抑制剂)抑制脂肪酸氧化仅对所有四种基因型细胞的生长有轻微影响。
使用RNA测序(RNA-seq)来探索这些细胞中不同代谢依赖性的遗传基础,提示这些BAF3-RTK细胞系中存在显著差异的基因簇,对这些显著基因簇进行KEGG通路富集分析,显示FGFR1和EGFR激活细胞中糖酵解和丝氨酸合成通路过表达(Fig. 1j)。分析EGFR和FGFR激活肿瘤中的代谢基因以探测BAF3细胞转录变化的临床相关性,差异代谢基因的KEGG通路富集分析(FC大于1.5倍; p <0.01)表明,富集到FGFR扩增肿瘤中的丙酮酸代谢和EGFR突变瘤中的甘氨酸丝氨酸和苏氨酸代谢,这些发现与BAF3细胞中鉴定的基因组结果吻合。这些数据共同证明了由癌细胞中相应的RTK活化驱动的代谢表型重编程的偏向性。
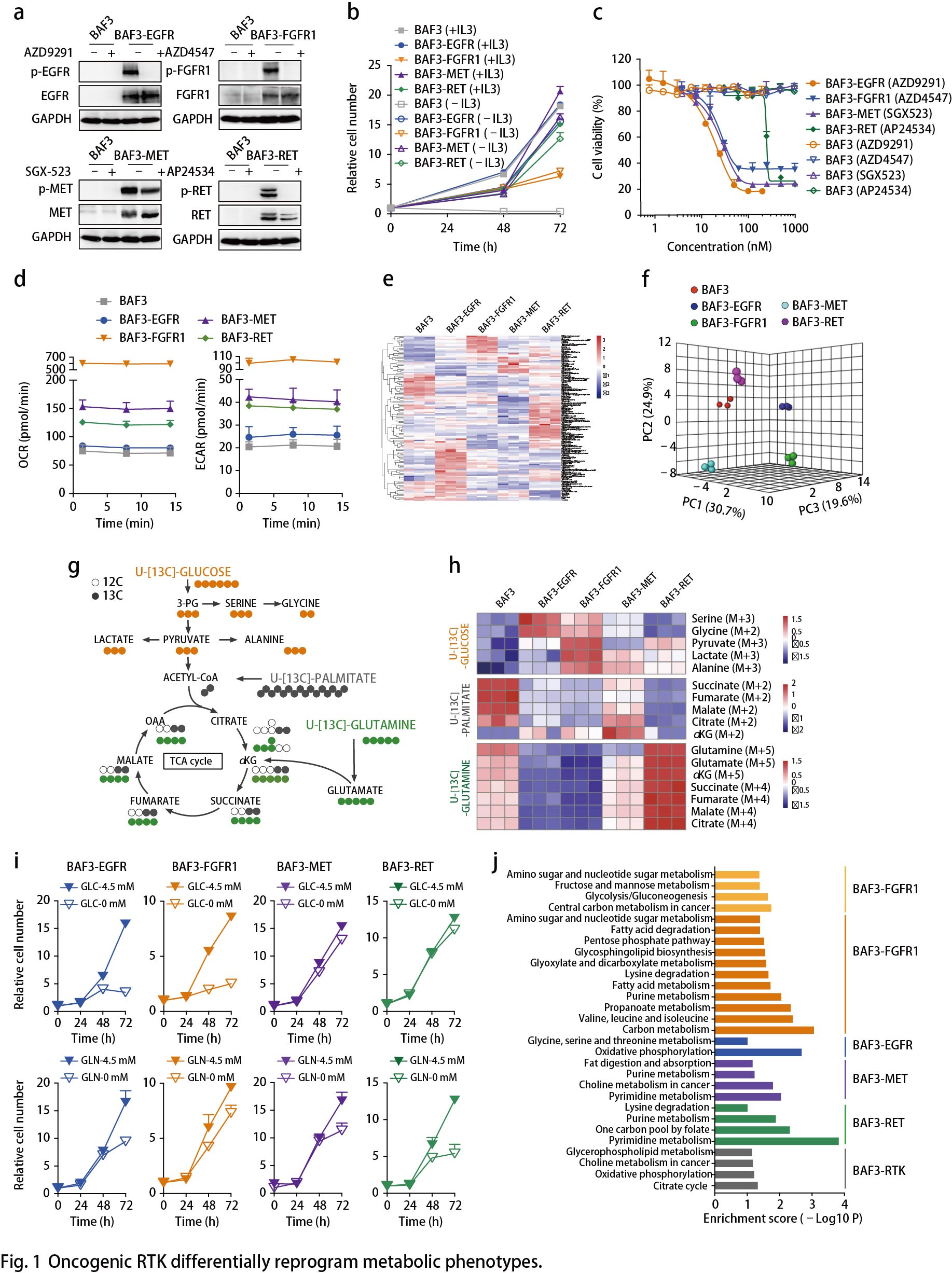
EGFR活化促进丝氨酸合成
以上数据表明EGFR活化后将糖酵解分支到丝氨酸合成通路(SSP)(Fig. 1h)。通过比较EGFR突变的癌症患者(n = 25)与野生型EGFR肿瘤(n = 715)(图2a),从TCGA数据库中提取的740名肺腺癌患者中进一步证实了上调的丝氨酸代谢,提示SSP可能是这种癌症亚型的临床相关易感性。用CBR5884(一种PHGDH抑制剂)处理RTK驱动的细胞以降低这些细胞中丝氨酸的从头合成,结果显示,BAF3-EGFR细胞对PHGDH抑制有反应,而BAF3-FGFR1细胞对PHGDH抑制无反应(Fig. 2b)。使用NCT503(另一种PHGDH抑制剂)观察到类似的结果。将具有FGFR1 / 2/3基因扩增的癌细胞和无驱动基因改变的野生型肺癌细胞(A431除外)两者均作为对照进行测试。使用CBR5884抑制PHGDH,EGFR突变细胞的生长抑制率高于FGFR突变细胞,野生型细胞大多无反应(Fig. 2c)。体内实验同样证实了这种代谢弱点:PC9移植模型(Fig. 2d)和具有EGFR L858R突变的NSCLC患者异种移植(PDX)模型LU-01-0251(Fig. 2e)用EGFR抑制剂Gefitinib或NCT503(唯一的PHGDH体内抑制剂)治疗,NCT503治疗后肿瘤生长受到显著抑制,略低于Gefitinib的EGFR抑制作用(Fig. 2d,e),但毒性最小。这些结果表明,EGFR驱动的癌症代谢弱点导致SSP依赖性。最近的研究还报道了SSP参与EGFR抑制剂的耐药性,从不同的角度证实了SSP在EGFR突变型癌症中的重要作用。
接下来作者探究丝氨酸生成的增加如何促进EGFR依赖性癌细胞的恶性生长。使用13C标记的葡萄糖作为示踪剂,BAF3-FGFR1细胞作为对照,检测BAF3-EGFR细胞中13C标记的核苷酸同位素异构体的贡献百分比(Fig. 2f),EGFR驱动细胞中M6-M9核苷酸同位素的比例增加,这表明甘氨酸和甲酸碳结合到嘌呤核苷酸中(Fig. 2f,g)。除核苷酸合成外,葡萄糖衍生的丝氨酸可通过甘氨酸的产生掺入谷胱甘肽(GSH)中,有助于氧化还原稳态。同时,13C标记的葡萄糖示踪LC-MS谱检测到葡萄糖衍生的GSH(M2同位素)也通过激活BAF3细胞中的EGFR而增强。与FGFR1扩增的DMS114细胞相比,敲低PHGDH或磷酸化丝氨酸磷酸酶(PSPH)使得EGFR突变的PC9细胞中的活性氧(ROS)增加。这些结果表明EGFR通过促进SSP来为DNA / RNA合成提供核苷酸。与FGFR1活化细胞相比,EGFR活化细胞相对较少依赖于丝氨酸的外源供应(Fig. 2h)。
EGFR活化究竟如何优先激活SSP?通过比较BAF3-EGFR和BAF3-FGFR1细胞之间SSP相关酶的表达,发现在EGFR活化后PSPH、丝氨酸羟甲基转移酶1(SHMT1),SHMT2和亚甲基四氢叶酸脱氢酶1(MTHFD1)等关键代谢酶特异性上调(Fig. 2i)。在同一组肺腺癌患者样本中观察到PHGDH和PSPH的显著上调(Fig. 2j)。通过具有突变EGFR(n = 6)或野生型RTK(n = 6)的NSCLC PDX肿瘤组织中对这些代谢酶进行免疫组织化学分析,结果显示,与野生型肿瘤相比,EGFR突变体肿瘤表现出更高的PSPH和PHGDH表达水平。而且,PDX模型(LU-01-0251)中上调的PHGDH和PSPH表达可以通过EGFR抑制剂的治疗来逆转(Fig. 2e,k)。
与携带野生型EGFR的患者相比,EGFR突变肺癌患者中PSPH(丝氨酸从头合成的关键酶及肝脏中该通路的限速酶)显著增加(Fig. 2j)。接着,作者调查患者的PSPH状态(来自TCGA数据库的1144名患者),发现16%的EGFR突变亚型中PSPH扩增突变和EGFR激活突变(L858R点突变,外显子19缺失或外显子21缺失)同时发生(Fig. 2l)。由于两种基因的遗传变化同时发生,这类NSCLC患者的SSP显著增强,提示。PSPH可能是NSCLC中EGFR活化的靶点与此观点一致,敲低PSPH导致EGFR突变体PC9和NCI-H1975细胞的生长显著降低,类似于EGFR失活(Fig. 2m)。以上数据突出了SSP作为EGFR突变癌症中的代谢弱点。
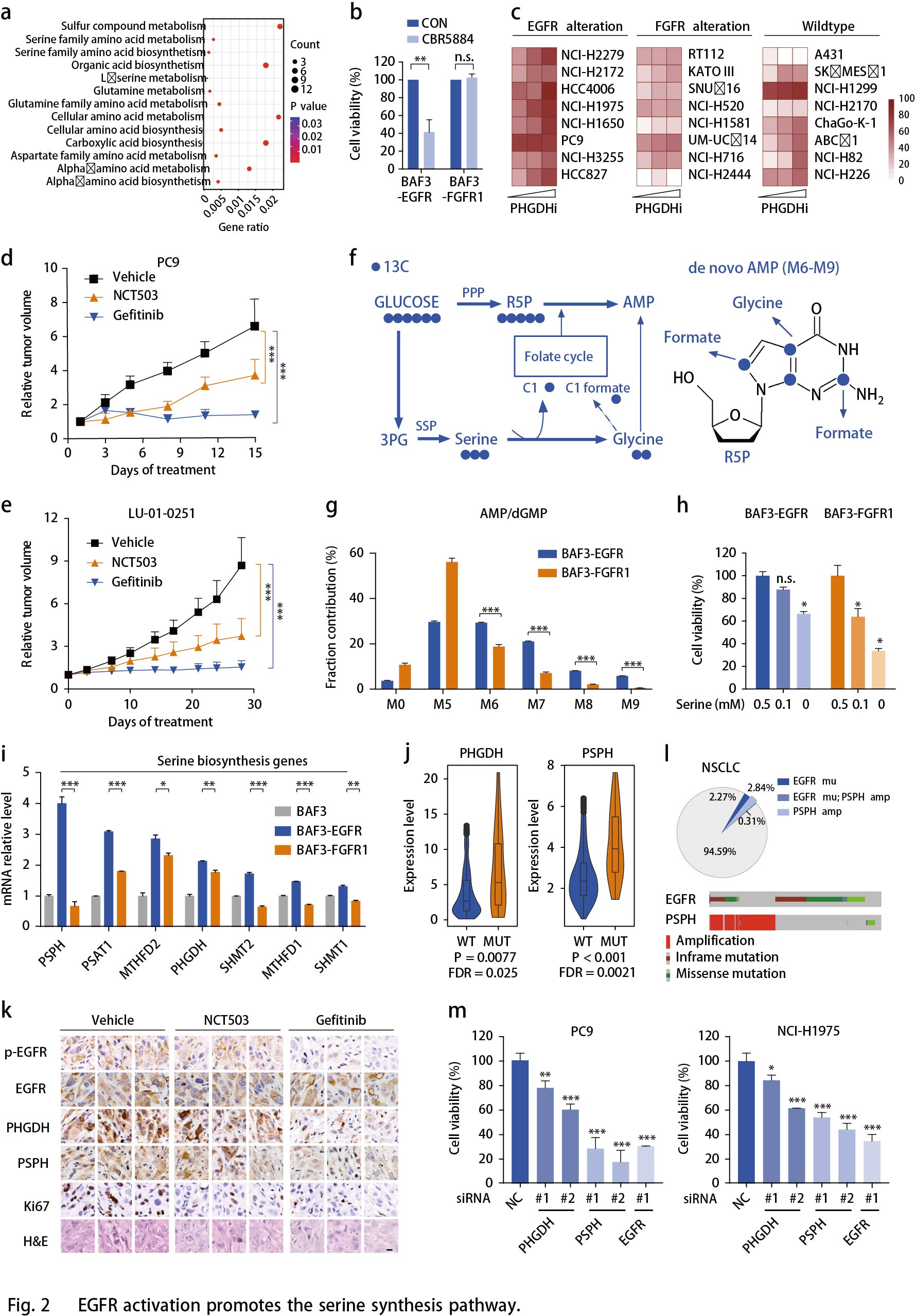
FGFR活化增强有氧糖酵解和乳酸循环
FGFR异常发生在多种类型的人类癌症中,包括NSCLC,膀胱癌和乳腺癌等。FGFR活化的细胞似乎消耗更多的葡萄糖进入糖酵解通路(Fig.1h),可从乳酸(糖酵解的关键产物)的胞内生成和胞外分泌的增强加以证实(Fig. 3a)。鉴于OCR的显著增强(Fig. 1d),作者推测乳酸可能作为TCA循环的替代碳源。为了支持这一假设,作者通过示踪13C标记的葡萄糖的碳富集到BAF3- RTK细胞中TCA循环的中间代谢物来检测葡萄糖对FGFR活化细胞中TCA循环中间体的贡献百分比(Fig. 3b)。在无标记葡萄糖(10mM)存在下,13C标记的乳酸(5mM)竞争性摄取分析显示FGFR细胞更倾向于消耗乳酸盐用于TCA循环(Fig. 3c)。为了追踪FGFR1扩增的NCI-H1581异种移植模型中葡萄糖和乳酸对TCA循环的贡献百分比,将13C标记的葡萄糖和13C标记的乳酸共同静脉注射小鼠30分钟后收集肿瘤组织,经外周转化后的血浆乳酸M3或M1同位素强度归一化后,掺入TCA循环中间体的葡萄糖碳与乳酸碳水平相当(Fig. 3d)。这些结果表明乳酸可以作为TCA循环中与葡萄糖同等的能源。与这些结果一致,使用Oxamate或GSK2837808A29抑制乳酸生成使得FGFR1扩增的NCI-H1581细胞中的OCR水平降低,类似于抑制FGFR的影响(Fig. 3e)。相反,乳酸的产生仅略微影响EGFR依赖性PC9细胞中的线粒体容量。
已知加速氧化磷酸化为恶性肿瘤生长提供ATP, 生成大量ROS副产物。检测乳酸对FGFR活化细胞中ATP和ROS产生的贡献,结果表明,在BAF3细胞中,与EGFR激活相比,FGFR1的活化与更高水平的ATP和ROS产生相关(Fig. 3f,g)。FGFR1激活的NCI-H1581细胞中ATP和ROS的产生均可被乳酸脱氢酶(LDH)抑制剂部分逆转,其程度与FGFR抑制相似(Fig. 3f,g)。总之,乳酸在促进FGFR异常癌的氧化磷酸化中起重要作用。
作者还比较了BAF3细胞中FGFR1或EGFR活化时糖酵解酶的表达。发现一些糖酵解酶如乳酸脱氢酶A(LDHA),ATP依赖性6-磷酸果糖激酶(PFKL),葡萄糖转运蛋白1(GLUT1)和己糖激酶2(HK2)在BAF3-FGFR1细胞中显著上调(Fig. 3h)。从TCGA数据库中提取的740例肺腺癌患者样品中证实了FGFR扩增与这些酶的上调相关(Fig. 3i)。使用免疫组织化学分析显示,相比RKT野生型肿瘤组织,FGFR1/2扩增的NSCLC PDX肿瘤组织中LDHA和HK2表达上调(Fig. 3j)。此外,作者使用基于CRISPR / Cas9筛选数据的Project Achilles来揭示细胞生长对这些代谢基因的依赖性。结果表明FGFR扩增的癌细胞中LDHA,PFKL和PKM的依赖性显著高于FGFR野生型癌细胞。作为对照,对PHGDH和PSPH的依赖性结果显示各亚组之间无差异,表明FGFR扩增细胞高度依赖于糖酵解。
所有这些发现都表明了乳酸在FGFR驱动的细胞中的重要作用。使用GSK2837808A(高选择性的LDHA抑制剂)抑制Fig. 2c所述癌细胞中的乳酸产生,与对照细胞相比,FGFR异常细胞的生长对LDH抑制剂更敏感(Fig. 3k),表明FGFR驱动的恶性生长优先需要乳酸代谢。在SNU16和NCI-H1581异种移植模型中乳酸抑制剂Oxamate显著抑制肿瘤生长且小鼠体重未发生变化(Fig. 31);13C葡萄糖示踪显示肿瘤中乳酸和柠檬酸生成降低(Fig. 3l)。在两个FGFR2扩增的NSCLC PDX模型中检测Oxamate的效果,结果显示在LU6429模型中,Oxamate或AZD4547的疗效都显著,但不同肿瘤之间疗效不同。在肿瘤生长抑制中,使用AZD4547-Oxamate组合对LDH和FGFR的组合抑制比单独的FGFR抑制效果更普遍且持续时间长(Fig. 3m),提示可以降低FGFR抑制剂的耐药性。小鼠体重变化提示组合研究具有良好的耐受性。在另一个FGFR2扩增模型LU0743中也观察到类似的趋势。同时,使用二甲双胍阻断线粒体呼吸,由于回收的乳酸被用于促进氧化磷酸化,与抑制乳酸生成相比,二甲双胍表现出相似的效果(Fig. 3m)。总之,这些结果表明在FGFR扩增的癌症中乳酸生成和氧化磷酸化的重要性,并提示由这种代谢表型产生的治疗前景。
前面结果表明,EGFR和FGFR基因的改变可以分别用于对SSP和乳酸产生抑制剂有反应的肿瘤进行分层。为了强化这一结果,作者还测试了携带野生型RTK的肿瘤模型。A431细胞异种移植常被用作RTK研究的对照,但PHGDH或LDH抑制剂几乎没有响应。同样,在无驱动基因改变的3种NSCLC PDX模型中(LU-01-0393,LU2071和LU-01-0416),未观察到明显治疗效果。这强调了患者选择这些抑制剂的重要性。
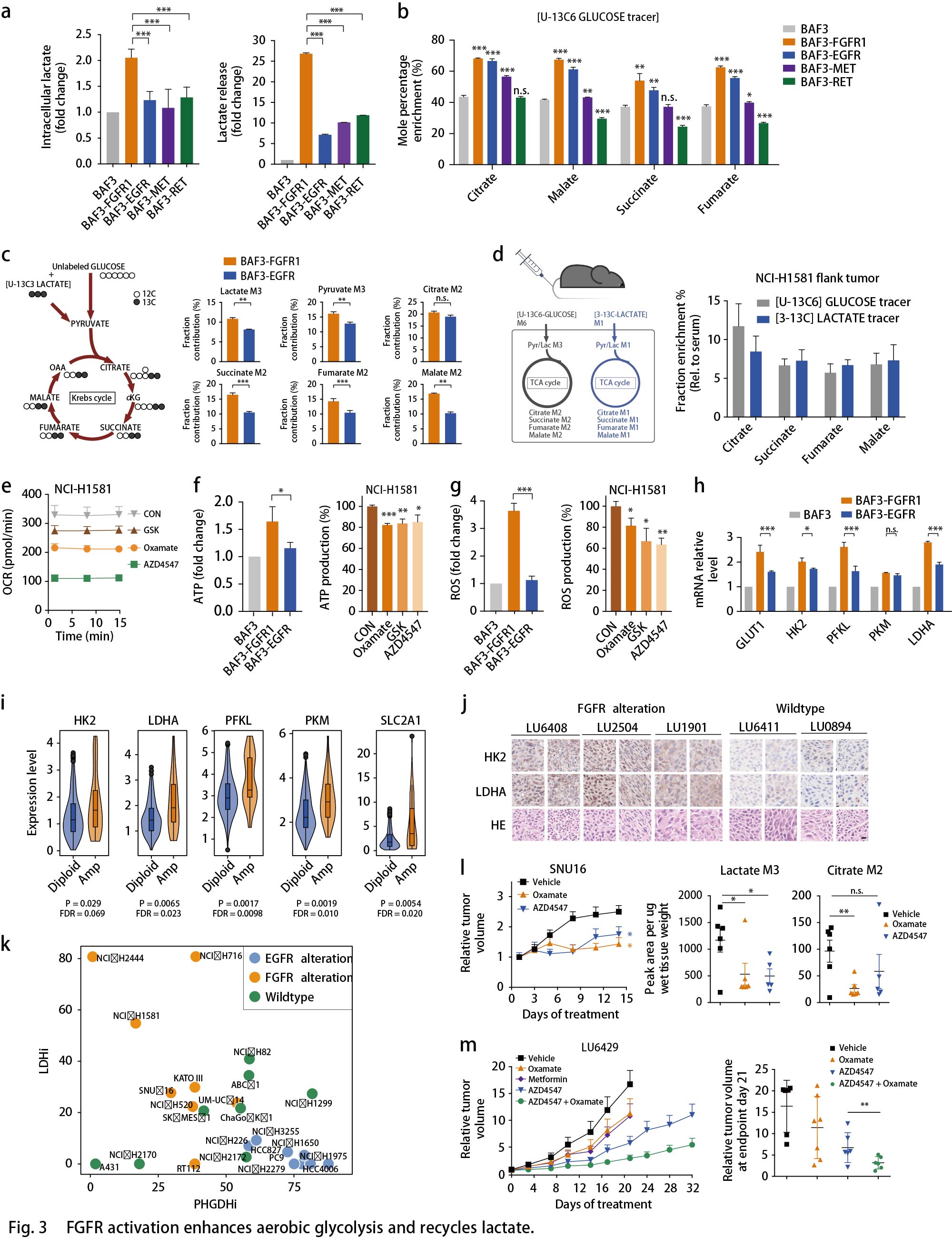
ATF4和MYC调控代谢重编程
剩下的问题是EGFR和FGFR细胞的优先代谢重编程?先前的证据突出了转录因子(TFs)作为癌细胞中代谢网络重编程的关键节点作用。作者探究FGFR和EGFR依赖性细胞中转录组重编程的TF。根据BAF3细胞中的RNA-seq数据,对RTK激活的代谢基因进行了分层(由KEGG数据库注释),并根据已发布的TF数据库(Cistrom,ORegAnno,mSigDB,CellNet和UCSC)建立描述TF-靶标互作的网络模型,分别获得了127个TF参与调节EGFR和138个TF参与调节FGFR依赖性差异转录的代谢酶。通过生物信息学注释进行功能性筛选:根据TF-靶标互作网络, 在FGFR3依赖的RT112细胞和EGFR激活的PC9细胞中,敲低25个代表性TF,并通过RT-qPCR分析检测SSP中PSPH,PHGDH的表达,以及糖酵解中LDHA,GLUT1的表达。最终在FGFR依赖性癌细胞中筛选出的ATF4,FOXO1,HIF1A,MAF,MYC和SREBP1,在EGFR依赖性癌细胞中筛选出的ATF4和MYC,分别是糖酵解和SSP中特征基因转录所必需的(Fig. 4a)。在这些TF中,只有HIF1A和MYC表达受FGFR抑制的显著影响。同样地,EGFR抑制剂Gefitinib可以影响EGFR突变癌细胞(Fig. 4b)和移植模型(Fig. 4c)中的ATF4和MYC表达。此外, MYC而非ATF4的敲低显著抑制FGFR3活化的RT112细胞的生长;而在EGFR异常的PC9细胞中,ATF4的敲低显著抑制细胞生长(Fig. 4d)。这些数据表明,HIF1A-MYC(在FGFR依赖细胞中)和ATF4-MYC(在EGFR活化细胞中)在转录调节代谢网络中发挥着至关重要的作用,MYC和ATF4分别在FGFR依赖的细胞和EGFR活化细胞中发挥着更为主导的作用。
通过网络分析来验证TF对在调节代谢网络中的密切关联,结果显示,有10个代谢基因由EGFR细胞中的MYC和ATF4共同调节,其中包括PSPH;20多个代谢酶基因由FGFR细胞中的HIF1A和MYC共同调控,其中包括LDHA(Fig. 4e)。仔细剖析这些TF对在两种细胞环境中的作用,发现敲低MYC下调HIF1A表达,而敲低HIF1A时MYC不变,证明MYC在FGFR细胞中的主导作用;同样的,在EGFR活化的PC9细胞中,敲低ATF4下调MYC表达,而敲低MYC时ATF4不变,证明ATF4在EGFR细胞中的主导作用(Fig. 4f)。接着,探究这些TFs对代谢表型的影响。与上述结果一致,FGFR活化的癌细胞中丙酮酸和乳酸的胞内生成通过MYC降低,而不是通过ATF4消耗而降低(Fig. 4g)。同时,在13C标记葡萄糖的ISA测定中,敲低ATF4而非MYC显著抑制PC9细胞中丝氨酸和核苷酸的合成,类似于EGFR抑制(Fig. 4h)。综上,作者建立了RTK驱动的转录调控网络,并突出了在EGFR或FGFR突变的癌症中,转录因子ATF4和MYC是代谢网络优先重编程的关键节点(Fig. 4i)。
小结
本研究采用代谢组学和转录组学相结合的方法来识别RTK驱动的癌症代谢弱点,建立了代谢弱点与癌症基因型之间的联系,确定了RTK异常的癌症代谢偏向性,提示RTKs的改变可用于代谢抑制剂的患者分层。FGFR致癌信号主要集中在MYC上,用于下游糖酵解酶的转录调控,而EGFR激活优先利用ATF4来驱动代谢网络重编程。总之,本研究确定了糖酵解和由此产生的乳酸的关键作用,它为FGFR异常癌症的能量生成提供TCA循环,并为EGFR组成性激活癌的核苷酸生物合成和氧化还原稳态的丝氨酸合成提供燃料。本研究可以从两个方面推进当前对该领域的理解:一方面,致癌的RTK驱动的代谢重编程可能导致明显的代谢弱点,可用于癌症治疗。另一方面,代谢异质性是由转录因子介导的代谢基因表达差异造成的。这些发现对指导代谢抑制剂的患者分层具有重要的转化医学价值。
参考文献
Jin N et al., Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat Commun. 2019 doi: 10.1038/s41467-019-10427-2.
原文下载,请长按识别下方二维码

